

Abstract
Il cancro è considerato una malattia genetica dalla maggior parte degli scienziati moderni.
Tuttavia, le prove suggeriscono che la disfunzione metabolica è alla base della patogenesi del cancro. Questo articolo si propone di fornire un quadro chiaro dei meccanismi alla base di questa malattia, valutandone le prove a supporto.
Introduzione
Il cancro è una malattia causata dalla proliferazione anomala delle cellule che porta alla formazione di una massa maligna, chiamata tumore. Un tumore può essere benigno o maligno. La caratteristica primaria di un tumore maligno rispetto a quello benigno è la sua capacità di metastatizzare. La metastasi è il processo durante il quale le cellule del tumore primario si staccano e viaggiano verso altre parti del corpo, dove proliferano.
Esistono diversi tipi di cancro a seconda del sito originale di manifestazione del tumore. Ad esempio, il cancro al seno ha origine nella zona del seno e può metastatizzare ad altri organi [1].
Sembra che, nonostante la nostra comprensione del cancro sia apparentemente migliorata, l’incidenza del cancro sia in aumento.
Più specificamente, si stima che nel 2050 il numero di nuovi casi di cancro sarà superiore del 77% rispetto al 2022 [2]. La ricerca sul cancro non dovrebbe concentrarsi solo sulla spiegazione dei meccanismi della malattia, ma anche garantire che questi risultati si traducano in risultati clinici migliori.
Questo articolo si propone di esplorare la patogenesi del cancro e di scoprire se la causa della malattia è genetica o metabolica.
Il cancro come malattia genetica – La teoria della mutazione somatica
Il cancro è comunemente percepito dalla comunità scientifica come una malattia genetica. In che misura questa teoria è vera e da dove ha avuto origine? La teoria genetica del cancro ha avuto origine nel 1914 in seguito all’ipotesi di Theodor Boveri secondo cui il cancro è causato da una segregazione cromosomica anomala durante la divisione cellulare.
Tuttavia, Boveri si è concentrato sulla citogenetica dello sviluppo negli organismi modello e non sembra aver condotto esperimenti diretti sul cancro. Sebbene Boveri si concentrasse sull’instabilità cromosomica alla base della mutagenesi, le sue osservazioni furono estese alle mutazioni somatiche, dando origine alla “teoria della mutazione somatica” del cancro [3].
Questa teoria è accettata dalla maggior parte degli scienziati moderni, anche se alcuni sostengono che dovrebbe essere riconsiderata [4]. Uno dei primi scienziati a mettere in discussione la teoria genetica del cancro è stato CD. Darlington. Ha proposto che il cancro derivi da mutazioni negli elementi citoplasmatici che ha chiamato “plasmageni” [5].
Le mutazioni somatiche sono i fattori trainanti della tumorigenesi cellulare?
Sebbene le cellule tumorali presentino mutazioni, non è chiaro se queste siano le cause della malattia o semplicemente l’effetto di una proliferazione incontrollata. Per indagare ulteriormente se gli elementi nucleari siano responsabili della tumorigenesi, sono stati condotti esperimenti di trasferimento citoplasmatico/nucleare [6].
Ciò ha rivelato incongruenze nella teoria delle mutazioni somatiche. Più specificamente, Israel et al. hanno trasferito il citoplasma dalle normali cellule epatiche epiteliali di ratto alle cellule tumorigeniche e hanno dimostrato che la tumorigenicità era soppressa in quattro cloni di progenie su cinque, come riassunto nella Figura 1. Ciò indica che gli elementi citoplasmatici hanno la capacità di controllare il destino delle cellule tumorali.
Inoltre, Mintz et al. hanno iniettato cinque cellule di teratocarcinoma derivate da embrioni nelle blastocisti e successivamente ne hanno osservato lo sviluppo [7]. Hanno concluso che le cellule cancerogene mostrano totipotenza evolutiva e hanno la capacità di generare tessuti adulti normalmente funzionanti. Questi risultati, tra gli altri, indicano che l’acquisizione della tumorigenicità non comporta cambiamenti nella struttura del genoma ma piuttosto cambiamenti nell’organizzazione dei tessuti.
Le prove a sostegno della teoria delle mutazioni somatiche si basano principalmente sul concetto di “mutazioni driver”. Le mutazioni driver sono definite dal National Cancer Institute come “cambiamenti nel DNA, sequenze di geni che fanno sì che le cellule diventino cellule cancerose e crescano e si diffondano nel corpo” [8]. Tuttavia, rilevare le mutazioni driver è un processo “notoriamente difficile” come riportato da Brown et al. [9].
Il processo di rilevamento di queste mutazioni prevede lo screening di ampie popolazioni di cellule tumorali per individuare mutazioni ricorrenti e la valutazione della loro mutabilità. Le mutazioni del conducente sono state associate a tassi di mutabilità inferiori rispetto alle mutazioni del passeggero (considerate il risultato dell’iperproliferazione tumorale e dell’instabilità genomica).
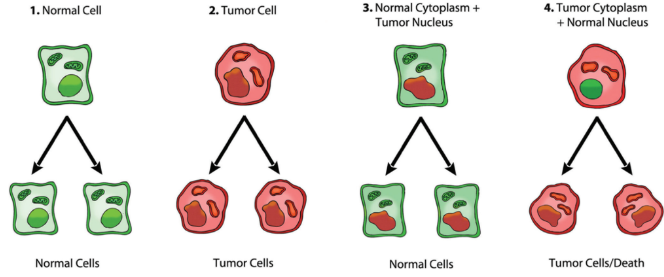
Figura 1: Riepilogo degli esperimenti di trasferimento nucleare/citoplasmatico che indicano che un nucleo tumorale combinato con un citoplasma normale può dare origine a cellule normali mentre un citoplasma tumorale con un nucleo normale può indurre tumorigenicità. Figura di Jeffrey Ling e Thomas N. Seyfried [10].
La limitazione introdotta da questo metodo è che mancano un gold standard oggettivo e criteri per definire una mutazione come driver [11]. Inoltre, studi sulla sequenza genomica hanno rivelato
che ci sono oltre 60 milioni di geni mutati espressi in diverse cellule tumorali, rendendo così impegnativo il compito di sviluppare terapie efficaci [12,13].
Inoltre, sebbene le mutazioni possano essere comuni tra le cellule tumorali, non esiste una singola mutazione genetica che possa essere riscontrata in tutte le cellule tumorali [12]. Ciò evidenzia la maggiore variazione tra
genomi tumorali e ci porta a considerare altri meccanismi patogeni.
Nel complesso, sebbene la teoria della mutazione somatica sia ampiamente accettata, è evidente che presenta incongruenze.
Questi ci spingono ad approfondire l’idea che gli elementi citoplasmatici/metabolici potrebbero essere i motori della tumorigenesi cellulare [10].
Il cancro come malattia metabolica
Otto Warburg fu il primo scienziato a descrivere la disfunzione metabolica nelle cellule tumorali e gli fu assegnato il premio Nobel per la fisiologia o la medicina nel 1931 [14]. Ha descritto che le cellule tumorali utilizzano anaerobici
glicolisi per generare energia anche in condizioni in cui l’ossigeno era abbondante [15].
Ha anche ipotizzato che questa via metabolica sia un adattamento della cellula a una respirazione insufficiente [16]. La via principale che viene influenzata durante la produzione di lattato è la fosforilazione ossidativa, che avviene nei mitocondri [17]. Ciò indica che durante la patogenesi del cancro le disfunzioni dei mitocondri possono portare a una respirazione compromessa e alla sovraregolazione della via glicolitica [10].
La glicolisi anaerobica è sostenuta attraverso la produzione di oncometaboliti: metaboliti cellulari endogeni che aiutano nella crescita e nella proliferazione del tumore [18].
Questi oncometaboliti possono anche agire come fattori di trascrizione, alterando l’espressione genica e inducendo la formazione di tumori [19].
Le prove sopra menzionate suggeriscono che la disfunzione metabolica precede l’instabilità genetica, essendo essa stessa il motore dell’alterazione dell’espressione genica.
Conclusione
Tenendo presente quanto sopra, si può concludere che esistono numerose prove a sostegno del fatto che il meccanismo della patogenesi del cancro implica una disfunzione metabolica. Ciò è caratterizzato da disfunzione mitocondriale che comporta un aumento della produzione di oncometaboliti e la sovraregolazione della glicolisi anaerobica.
Tuttavia, una combinazione di fattori genetici e metabolici contribuisce alla crescita
e metastasi di tumori. Pertanto, gli studi futuri dovranno concentrarsi sulla traduzione di questi risultati in approcci terapeutici che tengano conto delle scelte di vita e promuovano la salute metabolica.
Referenze
- National Cancer Institute. What is cancer? [Internet]. National Cancer Institute. National Institutes of Health; 2021. Available from: https://www.cancer.gov/about-cancer/understanding/what-is-cancer
- World Health Organization. Global Cancer Burden growing, Amidst Mounting Need for Services [Internet]. www.who.int. 2024. Available from: https://www.who.int/news/item/01- 02-2024-global-cancer-burden-growing-amidst-mounting-need-for-services
- Hanahan D, Weinberg RA. The Hallmarks of Cancer. Cell. 2000Jan;100(1):57–70.
- Soto AM, Sonnenschein C. One hundred years of somatic mutation theory of carcinogenesis: Is it time to switch? BioEssays. 2013 Dec 10;36(1):118–20.
- Darlington Cd. The Plasmagene Theory of the Origin of Cancer. British journal of cancer [Internet].
1948 Jun 1 [cited 2024 Apr 29];2(2):118–26. Available from: https://www.ncbi.nlm.nih.gov/pmc/ articles/PMC2007624/?page=1 - Israel BA, Schaeffer WI. Cytoplasmic suppression of malignancy. In Vitro Cellular &
Developmental Biology: Journal of the Tissue Culture Association [Internet]. 1987 Sep 1 [cited 2024 Apr 29];23(9):627–32. Available from: https://pubmed.ncbi.nlm.nih.gov/3654482/ - Mintz B, Illmensee K. Normal genetically mosaic mice produced from malignant teratocarcinoma cells. Proceedings of the National Academy of Sciences. 1975 Sep 1;72(9):3585–9.8.
- https://www.cancer.gov/publication s/dictionaries/cancer-terms/def/driver-mutation [Internet]. www.cancer.gov. 2011. Available from: https://www.cancer.gov/publications/dictionaries/cancer- terms/def/driver-mutation
- Brown AL, Li M, Goncearenco A, Panchenko AR. Finding driver mutations in cancer: Elucidating the role of background mutational processes. Nussinov R, editor. PLOS Computational Biology. 2019 Apr 29;15(4):e1006981.
- Metabolic Disease: Implications for Novel Therapeutics. AACR Education book. 2013 Apr 5;2013(1):31–6.
- Ostroverkhova D, Przytycka TM, Panchenko AR. Cancer driver mutations: predictions and reality. Trends in Molecular Medicine. 2023 Apr;
- Gyamfi J, Kim J, Choi J. Cancer as a Metabolic Disorder. International Journal of Molecular Sciences [Internet]. 2022 Jan 21;23(3):1155. Available from: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC8835572/
- Forbes SA, Beare D, Gunasekaran P, Leung K, Bindal N, Boutselakis H, et al. COSMIC: exploring the world’s knowledge of somatic mutations in human cancer. Nucleic Acids Research. 2014 Oct 29;43(D1):D805–11.
- Otto Warburg | German biochemist | Britannica [Internet]. www.britannica.com. Available from: https://www.britannica.com/biography/Otto-Warburg
- Warburg O. On the Origin of Cancer Cells. Science. 1956 Feb 24;123(3191):309–14.
- Liberti MV, Locasale JW. The Warburg Effect: How Does it Benefit Cancer Cells? Trends in Biochemical Sciences. 2016 Mar;41(3):211–8.
- Cooper GM. The Mechanism of Oxidative Phosphorylation. The Cell: A Molecular Approach 2nd edition [Internet]. 2000 [cited 2022 May 13]; Available from: https://www.ncbi.nlm.nih.gov/books/NBK9885/#:~:text=Most%20of%20the%20usable%20energy
- Yang M, Soga T, Pollard PJ. Oncometabolites: linking altered metabolism with cancer. Journal of Clinical Investigation. 2013 Sep 3;123(9):3652–8.
- Chowdhury R, Yeoh KK, Tian YM, Hillringhaus L, Bagg EA, Rose NR, et al. The oncometabolite 2-hydroxyglutarate inhibits histone lysine demethylases. EMBO Reports [Internet]. 2011 May 1;12(5):463–9. Available from: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3090014/